
ALL BLOGS
FMEA - The process medical device startup founders need to know

Written by
Published on
Apr 9, 2025

The process medical device startup founders need to know to get ready for ISO 14971 & 21 CFR Part 820

MedTech startups move fast. But speed without safety is risky, especially when entering regulated markets like the U.S. (FDA) or EU (MDR).
This is where FMEA comes in—a structured way to think about how your product might fail and how you’ll prevent that before it ever touches a patient.
Understanding FMEA is not only a best practice; it’s a practical gateway into ISO 14971 (risk management for medical devices) and 21 CFR Part 820 (the FDA's quality system regulation). This article will walk you through what FMEA is, why it matters, how it aligns with these standards, and how to implement it effectively - even in a lean startup environment.
What is FMEA (and why it matters)?
Failure Modes and Effects Analysis (FMEA) helps teams identify:
- What could go wrong (failure modes),
- Why it could happen (causes), and
- What the impact would be (effects on users or systems).
In short: it’s your blueprint for safety and regulatory readiness. FMEA is one of the foundational tools expected in both:
- ISO 14971 – Risk Management for Medical Devices (Europe)
- 21 CFR Part 820 – FDA’s Quality System Regulation (US)
How We Approach It (Real-World Example)
During project delivery, we usually map out a clear process for risk analysis alongside product development.
We begin by breaking down how users will interact with the device: Sleeping, walking, charging it, syncing to their phone. Then we ask, what could go wrong at each step?
We gather people from different teams—hardware, firmware, app development—and list all potential failure modes. For example:
- Patch disconnects during sleep
- Overheats near the battery
- Misinterprets breath patterns as apnea
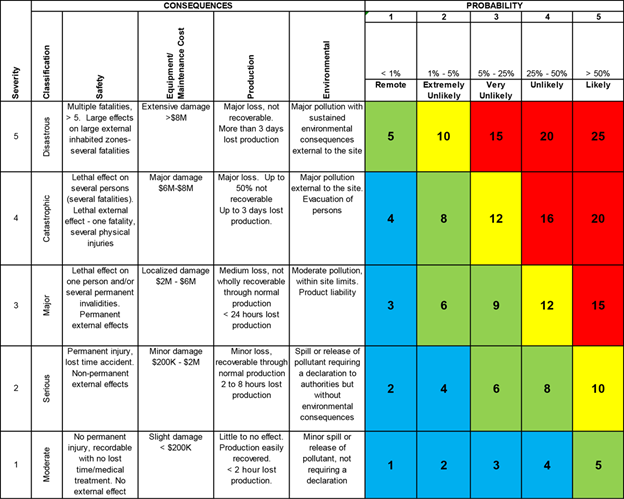
We then assess these risks by ranking each failure mode in terms of:
- Severity (how bad is the effect?),
- Occurrence (how likely is it to happen?), and
- Detection (how likely are we to catch it before harm occurs?).
The result: a Risk Priority Number (RPN) = Severity × Occurrence × Detection. That tells us where to focus. But the real value is in the discussion—founders start seeing risk as a design insight, not a roadblock.
Where FMEA Fits in ISO 14971 & FDA 21 CFR Part 820
Both standards expect you to show:
- You’ve identified risks across the entire product lifecycle
- You’ve designed controls to reduce those risks
- You’ve validated them through testing and real-world use cases
To be more specific:
- In ISO 14971:2019 – Clause 4 to 7, FMEA help systematically identifies hazards, analyzes risks, estimates severity/occurrence, and supports risk control measures
- In 21 CFR 820.30 (Design Controls), FMEA supports risk management as part of design validation and verification; can be linked to design inputs and outputs
- With Post-Market Surveillance, FMEA is a living document. It should be updated with field data, complaints, or CAPA (Corrective Actions) inputs
And that’s why FMEA is more than a form—it's a living part of your design control file.
Where to Apply FMEA in a MedTech Product
Depending on your product type, you may run one or more FMEAs, such as:
- System FMEA – on the whole device functionality
- Hardware FMEA – for embedded systems, sensors, PCB designs
- Software FMEA – for algorithms, cloud services, mobile apps
- Process FMEA – for your manufacturing or assembly workflows

Why Startups Need FMEA Early
Even if your device is still in prototype or software stage, you should start performing FMEAs as soon as your product has key functionalities or user interactions defined.
Why?
- Early Risk Awareness: FMEA helps catch design flaws before you invest in expensive testing, trials, or regulatory submissions.
- Compliance Alignment: ISO 14971 expects a structured process for identifying and controlling risks throughout the product lifecycle. FMEA is a standard tool to satisfy this requirement. 21 CFR Part 820.30(g) requires “design validation under defined operating conditions,” including risk assessment. FMEA is one of the acceptable methods.
- Investor and Partner Confidence: Documented FMEAs show diligence and maturity, especially when targeting regulated markets.
Common Mistakes I See
Here’s where founders slip up:
- Doing it after the design is locked
- Only involving engineers (no clinical input)
- Treating it like a checklist for investors or auditors
- Never updating it after product feedback or testing
A static FMEA is as risky as having none at all.

Late Failure Mode Discovery
How We Guide Our Clients Through FMEA in Early Stage
At ITRVN, during project delivery, we usually map out a clear process to help our startup partners handle FMEA even without an internal QA/RA team:
- Start from Use Cases: We break down real user scenarios—e.g., “patient wearing a patch ECG monitor while sleeping”—and map where failures could happen.
- Cross-Functional Brainstorming: We bring hardware, firmware, and software engineers into the room (or Zoom), because each team sees different risks. This ensures you don’t miss critical failure modes.
- Create the FMEA Table: Using templates aligned with ISO 14971, we log: The failure mode (e.g., “battery overheats”) Cause (e.g., “charging while in use”) Effect (e.g., “burn on skin”) S/O/D scores and calculated RPN
- Evaluate and Prioritize: We flag any RPN above a defined threshold (commonly 100 or more) and start mitigation plans. This may include software alerts, thermal sensors, or user instructions.
- Document and Iterate: FMEAs are not one-off tasks. We update them after test results, user feedback, or regulatory audits.
Best Practices for Startups Preparing for ISO 14971 & 21 CFR 820
- Use a Living Risk Management File – Your FMEA should be part of a central document that evolves from design to production to post-market.
- Link Risks to Design Controls – In your Design History File (DHF), make sure every high-risk area has a corresponding control (e.g., alarm, redundancy, fail-safe).
- Plan for Traceability – Especially in 21 CFR 820 audits, regulators will check that identified risks are tracked all the way to testing and validation stages.
- Automate Where Possible – Tools like Jama Connect, Greenlight Guru, or even structured Excel templates can help keep risk files organized and audit-ready.
Final Thoughts
FMEA isn’t just about compliance—it’s about building safe, reliable, and market-ready products. When done early and done right, it becomes a competitive advantage. For any startup eyeing the U.S. or EU healthcare markets, starting with FMEA is one of the smartest and most cost-effective decisions you can make.
If you're unsure how to begin or want to run a lightweight FMEA workshop for your MVP or alpha product, let’s talk. We’ve helped dozens of MedTech startups put scalable risk processes in place—long before they hired a full regulatory team.

If you’re not sure where to start, I’m happy to walk you through how we do it at ITRVN. No sales talk—just sharing what works so you don’t waste time figuring it out from scratch.
#medtech #fmea #startups #regulatory #iso14971 #quality #medicaldevices



